이건 첫번째 레슨, VUS 중 진짜 찾기 격주 목요일 오전 8시, 따끈따끈한 생물정보학 업계 소식이 당신을 찾아갑니다 |
|
|
안녕하세요, 생정해요 편집부의 파랑새 편집장입니다.
언질도 없이 장기간 휴재가 있었던 점 사과드립니다. 봄이 끝날때쯤 생정해요를 보내드렸는데, 벌써 여름이 끝나 가을이 목전입니다. 물건도 그렇듯, 어떤 일이든 마음이 무거울 수록 다시 시작하기가 어렵습니다. 어쩌면 생정해요의 휴재는 저에게 생정해요가 부담스러졌을때 부터 시작된 것 같습니다.
그럼에도 돌아왔습니다. 가벼운 마음으로 떠난 산책의 장점은 언제든 멈출수 있다는 것 뿐만 아니라, 언제든 다시 걷기 시작할 수 있다는 점입니다. 마음의 짐을 훌훌 털고, 초심으로 돌아가 보다 가벼운 어깨로 다시 산책을 나서려고 합니다.
가을이 왔습니다. 아니, 오고 있습니다. 구독자님은 가벼운 마음으로 시작한 일 중 잠시 멈춰둔 일이 있나요? 덮어두었던 소설책도, 미뤄두었던 일기도 좋습니다. 지리한 여름이 끝난 만큼, 파란 하늘 아래에서 함께 각자의 산책길을 떠나봅시다.
- 25년 9월 3일 날이 좋네요. 연구실에서, 파랑새 편집장 |
|
|
《Science (IF: 56.9)》
논문명: Deep generative models design mRNA sequences with enhanced translational capacity and stability
발간날짜: 2025/08/28
저자: He Zhang et al.
mRNA 백신은 COVID-19 대응에서 큰 성과를 거두었지만, 다른 질환으로 적용하려면 더 강력하고 오래 지속되는 단백질 발현이 필요합니다. 기존 연구들은 GC 함량이나 코돈 적합도 지수(CAI) 같은 단일 지표를 최적화하는 방식으로 접근했지만, 이는 전체 서열 맥락을 고려하지 못해 한계가 있었습니다. 최근 LSTM 기반 모델과 동적 프로그래밍 최적화 기법도 시도되었으나, 긴 서열 처리의 비효율성과 화학적 변형 mRNA에 적용 불가 등의 제약이 남아 있었습니다 .
이에 저자들은 언어 모델의 생성적 접근을 mRNA 설계에 접목한 GEMORNA라는 Transformer 기반 생성 모델을 개발했습니다. CDS는 단백질 서열을 “번역”하는 언어 모델로, UTR은 규칙성을 학습해 자유롭게 생성하는 시로 비유할 수 있습니다. GEMORNA는 코돈 사용, 구조 안정성, 자연스러움 점수 등 다양한 특성을 동시에 학습해 서열을 생성하며, in vitro 및 in vivo 실험에서 발현 향상을 입증했습니다. 예를 들어 firefly luciferase 발현은 기존 최적화 대비 최대 41배, EPO 발현은 최대 15배 증가했고, COVID-19 백신에서도 항체 역가가 크게 향상되었습니다. 또한 원형 RNA에서도 효율적인 발현을 보여 CAR-T 세포의 항종양 효과를 강화했습니다 .
이 연구는 mRNA 설계 문제를 단일 지표가 아닌 다차원적 최적화 문제로 보고, 생성 AI가 인간이 규칙화하기 어려운 서열 공간을 탐색할 수 있음을 보여줍니다. 생물정보학적으로는 언어 모델과 유전 부호 간의 평행성을 입증하며, 앞으로 백신 개발, 단백질 치료제, 유전자 치료 등 다양한 분야에서 범용적으로 활용될 수 있는 플랫폼을 제시했습니다.
|
|
|
《Genome Research (IF: 11.1)》
논문명: 3′-end ligation sequencing is a sensitive method to detect DNA nicks at potential sites of off-target activity induced by prime editors
발간날짜: 2025/09/03 (Online first)
저자: Jacob Stewart-Ornstein et al.
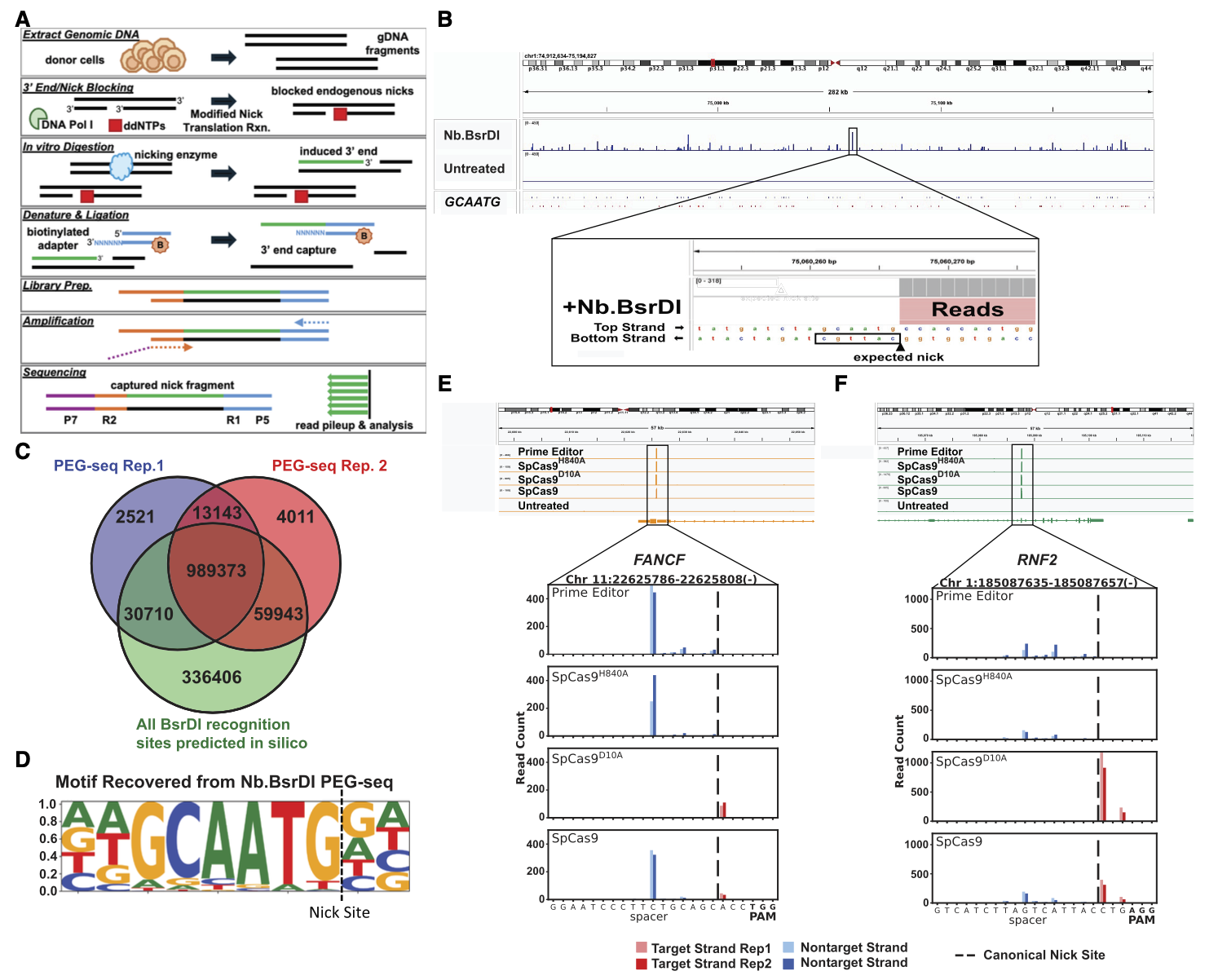
프라임 에디팅(prime editing)은 DNA의 한 가닥만 절단하는 새로운 편집 기술로, 기존의 이중가닥 절단(DSB) 기반 탐지법으로는 오프타겟 위치를 포착하기 어렵습니다. 기존의 CIRCLE-seq, CHANGE-seq 같은 방법들은 주로 SpCas9이 만드는 DSB를 검출하는 데 최적화되어 있어, 단일가닥 절단(SSB) 기반의 PE에는 한계가 있었습니다 .
저자들은 이 문제를 해결하기 위해 PEG-seq(3′-end ligation sequencing)을 개발했습니다. PEG-seq은 DNA nick(SSB)에 시퀀싱 어댑터를 직접 연결하여 효율적으로 잡아내는 방식으로, 기존 산화 손상 등 배경 신호를 차단하는 단계도 포함합니다. 이 방법은 Nb.BsrDI 같은 nickase 처리 DNA에서 70% 이상의 예상 절단 위치를 재현하며 높은 민감도를 보였습니다. 또한 SpCas9 변이체와 prime editor 단백질을 대상으로 한 실험에서, PEG-seq은 strand 특이적 절단 신호를 감지해 HNH와 RuvC 도메인의 차별적 활성을 분해능 있게 관찰할 수 있었습니다. 더 나아가, genome-wide 분석을 통해 수백~수천 개의 잠재적 오프타겟 nick 위치를 포착했으며, 이는 기존 방법으로는 파악되지 않던 수준의 민감도를 의미합니다 .
결국 PEG-seq은 프라임 에디터와 같은 nick 기반 유전자 편집 기술의 오프타겟 위험을 정밀하게 탐지할 수 있는 새로운 도구입니다. 생물정보학적으로는 오프타겟 데이터셋을 구축해 안전성을 정량화할 수 있으며, 임상 적용 전 편집 효소의 위험 프로파일링에 중요한 역할을 할 수 있습니다. 이는 유전자 치료제 개발에서 규제기관 평가와 안전성 검증에 핵심적인 방법론적 기여를 할 것으로 기대됩니다. |
|
|
《IEEE Transactions on Computational Biology and Bioinformatics (IF: 4.4)》
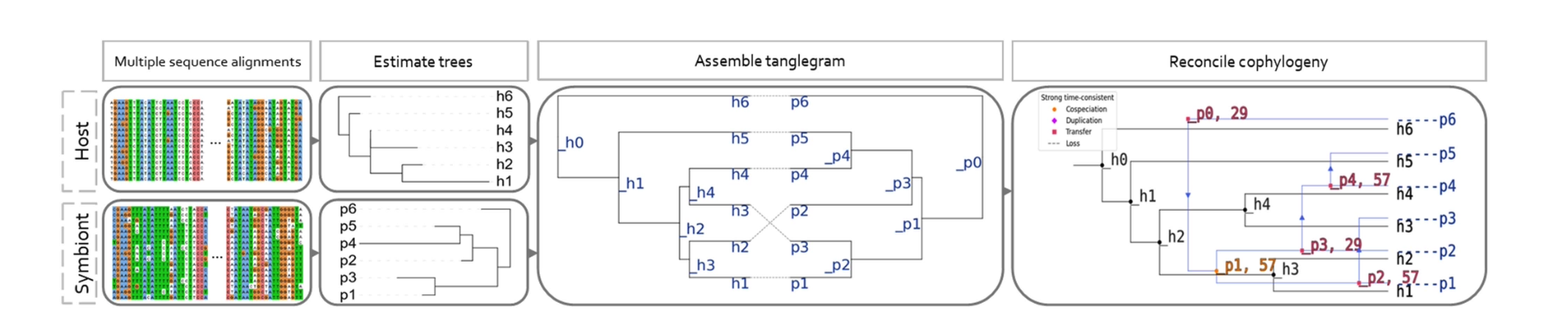
논문명: The Impact of Species Tree Estimation Error on Cophylogenetic Reconstruction
발간날짜: 2025/03/20 (current issue: 2025/08/08)
저자: Julia Zheng et al.
공진화(cophylogeny) 연구는 숙주와 공생체의 계통수 정보를 결합해 진화 역사를 추론하는 중요한 도구입니다. 그러나 대부분의 방법은 주어진 종 계통수(species tree)가 정확하다는 가정을 전제로 하지만, 실제로는 계통수 추정 과정에서 다양한 오차가 발생합니다. 이 오차가 이후 단계의 공진화 분석에 어떤 영향을 주는지는 체계적으로 밝혀진 바가 거의 없었습니다 .
저자들은 이를 규명하기 위해 시뮬레이션과 실증 데이터 기반의 대규모 분석을 수행했습니다. 혼합 시뮬레이션(mixed simulation)에서는 실제 데이터에서 추정된 계통수를 바탕으로 합성 데이터를 만들었고, 전방향 시뮬레이션(forward simulation)에서는 공진화 모델을 통해 계통수와 서열 진화를 함께 모사했습니다. 그 결과, 공진화 사건 추정 정확도는 계통수 추정 오류의 크기와 강하게 음의 상관을 보였으며, 다른 변수들(방법 선택, 이벤트 비용 설정 등)보다 훨씬 큰 영향을 미쳤습니다. 특히 phylogenetic error가 커질수록 cospeciation, host switch 같은 사건 분류의 정밀도가 급격히 떨어졌습니다 .
또한 연구팀은 두 가지 실증 시스템 (버섯균과 내생 세균, 오징어와 발광 세균) 을 분석하여 실제 데이터에서도 재현성이 낮아질 수 있음을 보여주었습니다. 이는 실험 설계 단계에서 종 계통 추정에 충분한 주의를 기울이지 않으면 잘못된 진화 해석을 내릴 위험이 크다는 것을 의미합니다. 생물정보학적으로 이 연구는 공진화 분석 파이프라인에서 종 계통수 추정의 정확성이 가장 중요한 변수임을 통계적으로 입증했으며, 향후에는 MSA 최적화, 샘플링 밀도, 추론 알고리즘 선택 등 upstream 단계의 엄밀성이 downstream 해석의 핵심임을 강조합니다.
|
|
|
박사 졸업은 끝이 아니라 시작.
'포닥은 늘고, 교수자리는 줄어든다'
지난달 28일 교육부가 발표한 통계에 따르면 국내 대학의 비전임 교원이 통계 작성이후 처음으로 15만명을 넘었다고 합니다. 작년 대비 올해 비전임 교원은 2.8% 늘어난 반면 전임 교원은 5% 가량 감소하였는데요. 대학의 재정난과 연구과제 축소와 맞물려 대한민국 이공계 인재들이 생계 절벽으로 내몰렸다는 말이 나옵니다.
HPP에 따르면 한국의 이공계 박사학위자는 인구 100만명당 199명으로, 미국 (130명)과 일본(96명)을 크게 웃도는 수준이지만, 이들의 일자리는 더더욱 감소하는 상황입니다. 결국 취직에 실패한 박사졸업자는 대학에 기댈수 밖에 없는 실정인데요.
대한민국 고등교육법 특성상 대학 구성원은 학생과 교직원으로만 나눠지면서 비전임 연구원들은 철저히 제도적 사각지대에 위치해 있습니다. 이들의 고용 안정성이 낮아진 만큼, 해외로의 인재 유출과 생계 절벽은 더욱 극단적으로 다가오고 있습니다.
|
|
|
유전체 데이터는 넘쳐나는데… 이 변이, 과연 병을 일으킬까요?
유전체 데이터는 쏟아지고, variant는 넘쳐납니다. 하루에도 수백 개씩 걸러지는 유전자 변이 중, 실제로 임상적으로 ‘의미 있는’ 변이는 얼마나 될까요? VUS(Variants of Uncertain Significance)는 여전히 판정을 미루는 회색지대에 남겨져 있고, 가족력 정보 없이 등장한 rare variant는 해석할 수 없다는 이유로 무시되기 일쑤입니다. 하지만 이제, 우리는 그 모호한 경계에 숫자를 붙일 수 있게 되었습니다. 그 변이가 질병을 ‘일으킬 가능성’을, 정량적으로 말입니다.
Mount Sinai의 연구진이 발표한 연구는, variant 해석의 패러다임을 “있다/없다”에서 “얼마나 자주 질병을 일으키는가”로 전환하는 접근을 제시합니다. 핵심은 penetrance, 즉 유전 변이 보유자가 실제로 질병을 발현할 확률을 예측하는 것입니다. 이는 단순한 유전자-질병 연관성을 넘어, 각 변이의 임상적 중요도를 실제 확률값으로 환산해내려는 시도입니다.
연구진은 미국 내 134만 명 규모의 EHR-연결 코호트에서 혈액검사, 심전도, 신장 기능 등 다차원 생체 데이터를 수집하고, 이를 기반으로 다중질환 예측 모델을 구축했습니다. 모델은 유전성 유방암(BRCA1/2), 가족성 고콜레스테롤혈증(LDLR), 다낭성 신장병(PKD1/2) 등 10개 주요 유전 질환에 대한 ‘질병 지수’를 학습하고, 이후 독립적인 엑솜 시퀀싱 코호트에 있는 1,648개 희귀 변이에 대해 AI 기반 penetrance 점수를 산출했죠.
무엇이 새로웠을까요? 이 penetrance 점수는 기존의 이진 분류(pathogenic vs. benign)보다 훨씬 섬세한 정보를 제공합니다. 같은 질병과 연관된 loss-of-function 변이라 하더라도, 어떤 변이는 실제 발현 확률이 70%에 육박한 반면, 다른 변이는 10% 미만이었습니다. 기존의 분류체계로는 같은 박스에 묶였을 이들 변이가, 이제는 전혀 다른 임상적 의미를 갖게 된 겁니다. 특히 새롭게 발견된 혹은 임상적으로 모호했던 VUS에 대해, ‘이건 병을 일으킬 가능성이 높은 VUS’라는 식의 계량적 판단이 가능해졌다는 점이 중요합니다.
실제 환자 데이터를 보면 더 흥미롭습니다. 높은 penetrance 점수를 받은 변이 보유자는, 질병 진단을 받지 않았더라도 관련 생체 지표가 이미 악화되어 있는 경우가 많았고, 진단 시점도 더 빨랐습니다. 즉, 모델은 단순히 과거의 진단을 재해석하는 데 그치지 않고, 미래의 질병 가능성을 조기에 포착하는 신호로 작동할 수 있는 셈입니다.
물론, 이 기술이 당장 모든 임상 현장에서 적용될 수 있는 것은 아닙니다. 훈련된 모델이 특정 인구집단(미국 기반 백인 중심 데이터)에 최적화되어 있다는 점, 환경 요인과 상호작용하는 다양한 유전자-질병 경로를 온전히 포착하기는 어렵다는 점도 한계입니다. 하지만 유전체 데이터 해석에서 가장 어려운 문제 중 하나인 ‘불확실한 변이’를 수치적으로 접근할 수 있다는 점은, 바이오인포매틱스와 정밀의료의 접점을 넓히는 중요한 진전입니다.
어쩌면 우리는 지금, 진짜 변이와 가짜 변이 사이에 서 있습니다. 두 변이 모두 데이터상에 ‘존재’하지만, 하나는 질병의 경고음이고, 다른 하나는 의미없는 잡음일 수 있죠. 그 둘을 구분해내는 건, 단순한 필터링이 아니라, 발현 가능성에 대한 ML적 접근입니다. 우리는 이제 ‘병을 유발하는 변이’를 찾는 것이 아니라, ‘어느 정도 병을 유발할 가능성이 있는 변이’를 판단하는 시대에 들어서고 있습니다.
변이는 존재하지만, 질병은 아직 오지 않았습니다. 이에 기반한 조기진단이 가져올 의료의 혁신이 기다려집니다.
해당 기고문은 25년 8월 국제 학술지 Science 에 실린 논문, "Machine learning–based penetrance of genetic variants" 에 기반하였습니다.
|
|
|
|