그것도 아주 오래전부터. 격주 목요일 오전 8시, 따끈따끈한 생물정보학 업계 소식이 당신을 찾아갑니다 |
|
|
안녕하세요, 생정해요 편집부의 파랑새 편집장입니다.
인류가 환경을 파괴하고 있다는 말은 아주 오래전부터 여러분들께 익숙한 사실일 것 입니다. 그런데 궁금한 것은, 그 시작이 언제부터였을까요? 우리가 자연의 일부에서 벗어나, 다른 생태계에 대해 비가역적 영향력을 미치기 시작한 그 시점 말입니다. 오늘의 요주의 논문은 제게 아주 인상깊었던 한편의 논문을 소개해드립니다. 이제 가을이 오고 있는 것 같습니다. 선선한 날씨 맘껏 즐기시며 평안한 하루 되시길 바랍니다.
- 25년 9월 17일, 비가 오네요. 라운지에서, 파랑새 편집장 |
|
|
《iMeta (IF: 33.2)》
논문명: fastp 1.0: An ultra-fast all-round tool for FASTQ data quality control and preprocessing
발간날짜: 2025/09/09
저자: Shifu Chen
FASTQ 전처리 도구 중 하나로 널리 사용되어온 fastp가 처음으로 메이저 업데이트를 맞았습니다. 이번에 발표된 fastp 1.0은 단순히 기능이 추가된 버전을 넘어, 대규모 데이터 시대에 최적화된 전천후(preprocessing + QC + 보고서 생성) 플랫폼으로 재정의되었다고 볼 수 있어요.
기존의 Trimmomatic, Cutadapt와 같은 도구들은 어댑터 제거, 필터링, 품질 보고서 생성을 별개로 처리해왔습니다. 이 때문에 유저는 여러 툴을 조합해야 했고, 리소스 낭비도 발생했죠. 반면 fastp는 모든 작업을 하나의 명령어로 처리하며, 특히 자동 어댑터 탐지, polyG/polyX trimming, UMI 처리, 중복 제거 등 최신 시퀀싱 장비에 최적화된 기능을 기본 제공합니다.
fastp 1.0은 HTML 기반의 전후 비교 리포트, 일괄 처리(batch processing), 효율적인 멀티스레드 구조, 그리고 자체적으로 재현 가능한 결과까지 모두 지원하며, 클라우드 환경에서도 경량으로 동작할 수 있도록 설계됐습니다. 실제 실험에서도 fastp는 Trimmomatic이나 Cutadapt보다 훨씬 많은 작업을 수행하면서도 3~6배 이상 빠른 성능을 보였습니다.
이번 업데이트의 핵심은 ‘기능의 많음’이 아니라, 사용자 관점에서 생물정보학 도구가 어떻게 설계되어야 하는지를 설득력 있게 제시했다는 점입니다. 파이프라인에서 가장 먼저 쓰이는 전처리 도구의 표준이 완전히 재정의되었다고 해도 과언이 아닙니다.
|
|
|
《Briefings in Bioinformatics (IF: 13.994)》
논문명: DeepMobilome: predicting mobile genetic elements using sequencing reads of microbiomes
발간날짜: 2025/08/08
저자: Youna Cho et al.
모바일 유전 요소(MGEs)는 항생제 내성 유전자나 병원성 유전자의 전파를 유도하는 주요 매개체로, 마이크로바이옴의 유전체 내에서 동적인 진화와 기능적 확산을 이끕니다. 하지만 실제 마이크로바이옴 환경에서는 다양한 균주와 복잡한 서열 조합 때문에 MGEs의 정확한 탐지가 어렵고, 기존 도구들은 대부분 단일 유전체 환경을 가정하고 설계되어 한계가 뚜렷했습니다.
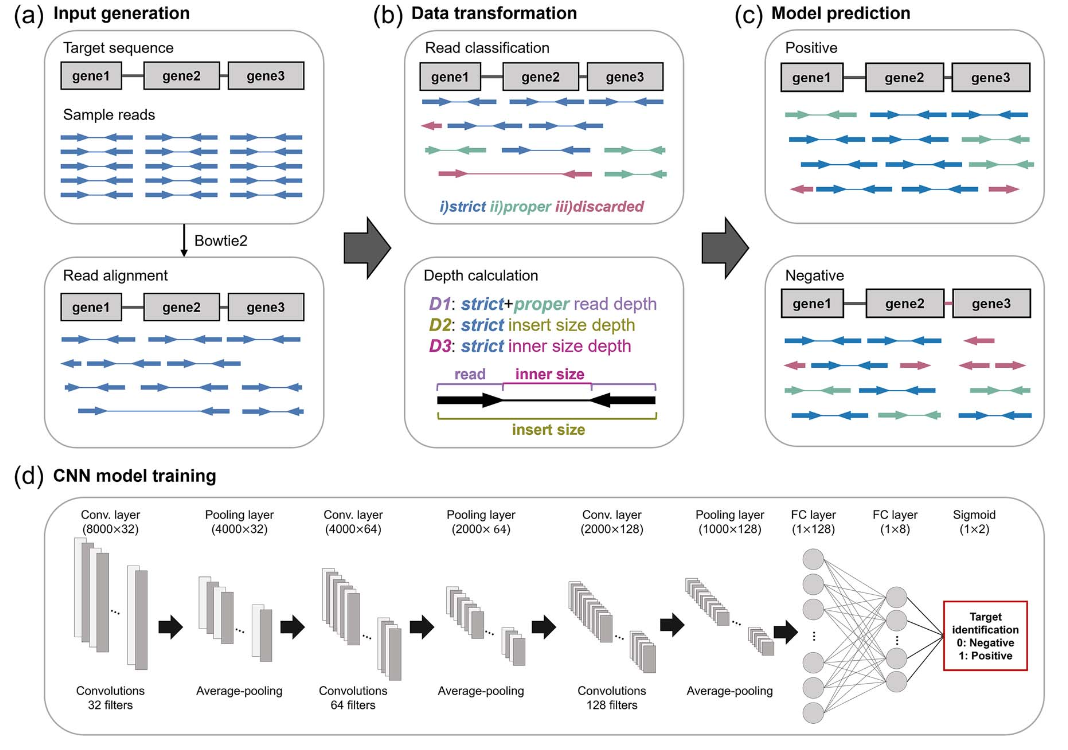
이 논문은 마이크로바이옴 내 MGEs를 정확하게 탐지할 수 있도록, read 정렬 정보를 이미지 형태로 변환한 후 CNN을 통해 예측하는 DeepMobilome 모델을 제안합니다. 기존 방식과 달리 단순히 서열 유사성을 비교하지 않고, read들의 정렬 유형(strict/proper/discarded), depth, insert size, inner size 등을 3채널 입력으로 구성해 위치 기반의 구조적 패턴을 학습합니다.
DeepMobilome은 실험적으로 단일 유전체, 마이크로바이옴 시뮬레이션, 실제 fecal sample 등 다양한 조건에서 테스트되었으며, 기존 도구(MGEfinder 등) 대비 20% 이상의 정확도 향상을 보였고, 마이크로바이옴 환경에서도 F1-score 0.939의 우수한 성능을 입증했습니다. 특히 구조적 비정형성을 반영할 수 있는 점에서 부분 유사한 MGE까지 정밀하게 검출 가능하다는 점이 강점입니다.
이 연구는 기존의 “alignment dependent MGE 탐지” 방식의 한계를 넘어, 구조 패턴 기반 딥러닝 분석이라는 새로운 방향을 제시했습니다. 항생제 내성 유전자 추적, 병원성 전파 경로 분석, 감염병 대응을 위한 마이크로바이옴 surveillance 등 다양한 응용 분야에서 실질적인 기여가 기대됩니다. |
|
|
《IEEE/ACM Transactions on Computational Biology and Bioinformatics (IF: 4.4)》
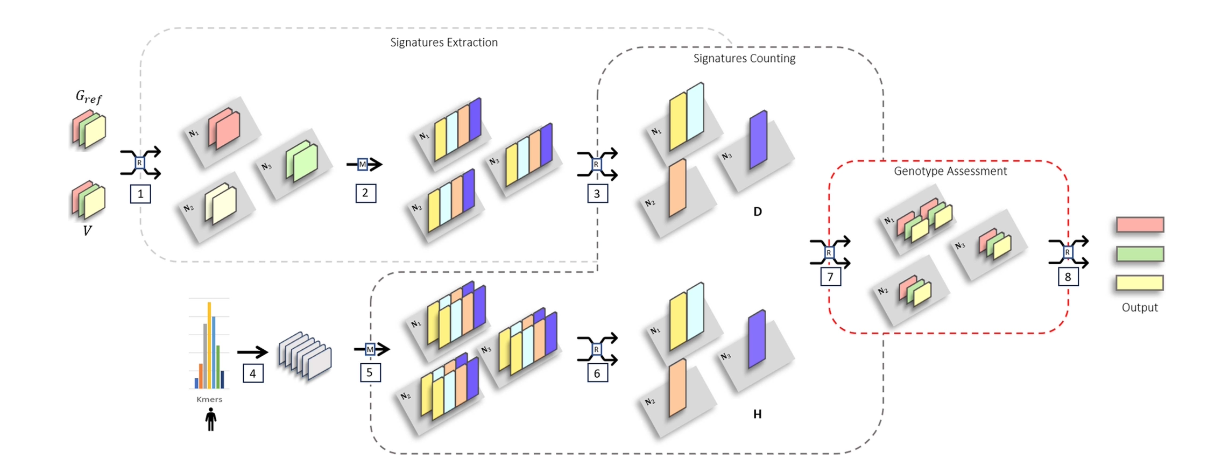
논문명: Efficient and Scalable Alignment-Free Distributed Genotyping of SNPs and Short Indels
발간날짜: 2025/01/09
저자: Lorenzo Di Rocco et al.
대규모 유전체 연구가 급증하면서 수천 명 규모의 변이(SNP 및 short Indel) 분석이 일상화되고 있지만, 여전히 주류 파이프라인(GATK, BWA 등)은 정렬 기반 접근 방식에 의존하고 있습니다. 이는 정확하긴 하지만 계산량이 많고, 클러스터 환경에서 병렬화가 어려우며, 효율성이 떨어지는 단점이 있습니다.
이 논문은 이러한 한계를 해결하기 위해 Distributed Alignment-Free Genotyping (DAG)이라는 새로운 시스템을 제안합니다. DAG는 전통적인 BAM 파일 기반 variant calling을 버리고, K-mer 기반의 alignment-free 전략으로 SNP 및 indel을 판별합니다. Apache Spark 기반으로 분산 환경에 맞춰 설계되어, 수십 TB의 데이터도 빠르게 처리할 수 있습니다.
중심 아이디어는 K-mer 기반으로 사전 구축된 변이 인덱스에서, 관찰된 read에서 변이를 유추하는 방식입니다. 여기에 quality filter, abundance threshold, paired-end 정보 등을 함께 고려하여 정확도 손실 없이 고속 처리가 가능하게 했습니다. 특히 10K 샘플에 대한 대규모 테스트에서도 precision과 recall이 98% 이상을 유지하면서 GATK 대비 최대 50배 빠른 처리 속도를 보였습니다.
이 연구는 단순히 속도만 빠른 것이 아니라, 변이 분석 파이프라인의 설계 철학 자체를 정렬 없는 방식으로 바꾸었다는 점에서 의미가 큽니다. 대규모 인구 유전체 연구, 병원 기반 변이 스크리닝, 마이크로바이옴 다양성 분석 등에서 확장성이 뛰어나고, 파이프라인 경량화가 필요한 저자원 환경에서도 유용하게 적용될 수 있습니다.
|
|
|
미국 포닥 들어와주세요
'해외 우수 연구자 1000명 유치 목표'
정부의 해외 이공계 인력 유입 사업인 이노코어 (InnoCORE) 사업을 내년 확장하겠다고 발표하였습니다. 관련 예산은 600억 규모로, AI 부터 첨단 바이오 분야에서 우수 해외연구자를 유입, 4대 과학기술원 (KAIST, GIST, UNIST, DGIST) 에 박사후 연구원으로 (...?) 채용하며, 연봉 9천만원과 인당 연구비 6천만원을 지원하겠다고 발표하였습니다.
(개인적으로는 국내 신진 연구자들의 기본적인 처우개선도 같이 진행되어야 하지 않을까 하는 생각입니다.)
|
|
|
“우리가 미치는 영향력”
인간은 오래전부터 자연을 바꾸며 살아왔습니다. 하지만 ‘어디까지가 변화이고, 어디부터가 파괴인지’를 판별하는 일은 여전히 쉽지 않아요. 특히 곤충처럼 작고, 오래된 기록이 부족한 생물들에게는 더더욱 그렇습니다. 최근 Science 지에 실린 한 연구는 이 질문에 생물정보학을 통해 답하고자 합니다.
이번 연구는 오키나와과학기술대학원(OIST), 하버드대, 호주국립대, 예일대 등 국제 공동 연구진이 수행했으며, 피지 제도에서 채집한 개미 4132마리를 RAD-seq 기반 ‘커뮤니티 유전체학’(community genomics)으로 분석한 결과입니다.
기존 곤충 감소 연구는 대부분 최근 수십 년간의 관측 자료에 의존해 왔습니다. 하지만 이 논문은 섬 생물 군집 전체의 진화사와 개별 종의 인구 변화 양상을 수천 년 전부터 지금까지의 시간 축으로 복원했어요. 핵심은 ‘현재 채집된 시료에서 유전 정보만으로 과거의 개체군 크기를 추정하는’ 접근이 가능해졌다는 것입니다. 이를 통해 연구진은 총 127종의 개미에 대해 고해상도 유전 분석과 계통 재구성을 시도했고, 그 결과 피지에서 독자적으로 진화한 고유종의 79%가 지난 수천 년간 인구 감소를 겪어왔다는 사실을 밝혀냈습니다.
흥미로운 점은, 감소 시점이 인간의 초기 정착(약 3000년 전)과 정확히 맞물린다는 점이에요. 특히 산림 개간과 농경 확대, 유럽인의 도래와 산업농업의 시작, 외래종 도입 등의 사건 이후 고유종 개체군의 감소 속도는 더욱 빨라졌습니다. 반면, 농경지·도시 등 교란 환경에 적응한 외래종과 태평양 주변의 광범위한 개체군은 오히려 빠르게 확산되며 번성했습니다.
이 연구의 생물정보학적 핵심은 단순히 “개미가 줄고 있다”는 관찰이 아니라, 유전체 기반으로 시간적·공간적 감소 패턴을 정량화하고, 진화적 맥락에서 그 원인을 분해했다는 점입니다. RAD-seq 기법을 통해 수천 개체의 유전 구조를 재구성한 뒤, 공동 조상으로부터의 이입 시간, 개체군 크기 변화, 서식지 교란 허용도 등 다양한 요인을 통합 분석했습니다. 특히 Bayesian phylogenetic 모델을 활용해 교란 민감도와 고도(지리적 분포) 같은 생태적 요소를 유전적 경향성과 연결시킨 부분은, 기존 ‘단일 종 관찰’ 방식과 뚜렷한 차이를 보입니다.
또한 중요한 함의는 ‘섬’만의 이야기가 아닐 수 있다는 점입니다. 연구진은 “고립된 섬에서 진화한 종이 교란에 취약한 건 맞지만, 본토에서도 서식지가 점점 섬처럼 조각화되고 있다”고 경고합니다. 고유종의 멸종은 이미 시작되었고, 유전체는 그 사실을 말없이 기록하고 있었습니다.
결국 우리는 숫자의 함정에 빠지지 말아야 합니다. 총 곤충 개체수나 생물량이 유지된다고 해서 생태계가 건강하다는 뜻은 아닙니다. 진화적으로 독립된 고유종이 사라진다면, 그 생태계는 구조적으로 무너지고 있는 것일 수 있어요.
이제 질문은 이렇게 바뀝니다. ‘변화에 적응한 종이 살아남는’ 자연의 법칙이, 인간이 만든 환경에도 적용되는 게 옳은 일일까요? 그렇게 선택된 자연은 과연 우리가 지키고 싶던 자연일까요? 구독자님은 어떻게 생각하세요?
해당 기고문은 25년 9월 국제 학술지 Science 에 실린 논문, "Genomic signatures indicate biodiversity loss in an endemic island ant fauna" 에 기반하였습니다.
|
|
|
|